细胞捕手
2026年3月16日,劳伦斯伯克利国家实验室及密苏里大学的研究人员在《Current Opinion in Structural Biology》上发表综述文章,题为“Artificial intelligence methods for protein structure and interaction prediction: Recent advances and challenges”。作者概述了以蛋白质为核心的生物分子结构与相互作用预测的近期进展,重点指出了该领域面临的一些主要挑战,并探讨了应对这些挑战的潜在方向。
结构生物信息学领域迎来了人工智能研究者的极大关注。从蛋白质结构与相互作用预测和设计(图1),到基于结构的药物开发,近期为解决基础生物学问题而引入的新型深度学习算法,正在开创一个数据驱动的计算结构生物学与生物物理学的新时代。文章重点选取了该领域当前及未来的一部分发展方向,旨在阐明研究已经走向何方、可能走向何方,或者在未来几年中需要走向何方。将从三个部分展开论述(蛋白质三级结构预测,蛋白质-蛋白质相互作用预测,以及蛋白质-其他生物分子相互作用与动力学),每一部分聚焦于基于AI的结构生物信息学的不同重点领域,最后以总结性思考与结论作为结束。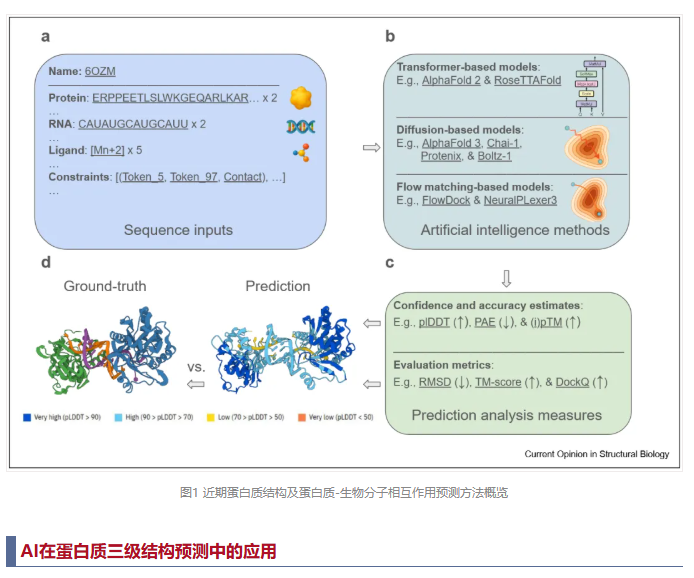
随着高精度三维构象深度学习方法的应用,蛋白质三级结构预测取得了飞速发展。基于Transformer的方法引领了这场革命,首先是AlphaFold 2引入了Evoformer模块共同建模序列关系和配对关系,为预测精度树立了新的标准。RoseTTAFold通过三轨架构整合一维、二维和三维数据,扩展了这一框架;而ESMFold和OmegaFold等无需多序列比对(MSA)的方法,利用大语言模型直接从单条序列预测结构。OpenFold、HelixFold和MassiveFold在AlphaFold 2的基础上进行了改进,优化了效率、模块化和可扩展性。同时,MULTICOM3和MULTICOM4等基于输入优化的方法通过改进MSA和模板选择提升了AlphaFold的精度;而AFsample和AF_unmasked利用dropout采样来探索模型的不确定性。trRosettaX2通过从输入序列中移除无序区域并整合残基间距离预测,提高了预测准确性。在2024年举行的第16届蛋白质结构预测技术关键评估(CASP16)国际竞赛中,MULTICOM4和trRosettaX2均位列顶级三级结构预测器之列。
除基于Transformer的架构外,基于扩散的方法已成为一种强大的生成式替代方案。AlphaFold 3率先采用全原子扩散模型,不仅能够模拟蛋白质单体,还能模拟复合物、核酸以及配体结合结构。HelixFold3、Chai-1、Protenix和 Boltz-1等开源实现进一步扩展了基于扩散的建模。基于流匹配的技术通过学习平滑、连续的变换来生成三级结构,提供了一种极具吸引力的替代方案,NeuralPLexer3便是其中的代表。总体而言,这些方法突显了深度学习驱动的结构预测领域日新月异的格局,Transformer、生成式模型以及混合模型不断推动着生物分子建模的发展边界。此外,EnQA和GCPNet-EMA等精度评估方法使得无需依赖MSA衍生的特征,即可快速评估预测蛋白质结构的质量。表1列出了近期用于预测和评估蛋白质三级结构的方法。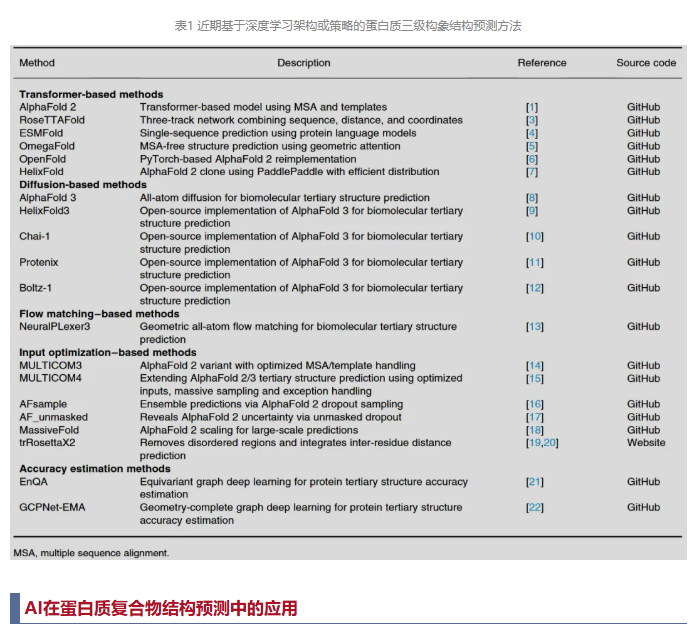
人工智能的进展已从根本上重塑了蛋白质复合物(即多链或多聚体)结构预测领域。在AlphaFold-Multimer等深度学习方法出现之前,大多数传统方法依赖于基于模板的建模或分子对接,这些方法主要聚焦于二元蛋白质相互作用,在处理更大的复合物时面临困难。AlphaFold-Multimer的发布标志着一个重要的转变,它建立了一个基准,连接了传统的以二聚体为中心的方法与能够准确预测含有多条链的复合物结构的现代方法。
如表2所示,近期用于蛋白质复合物预测的人工智能方法可根据其底层深度学习架构大致分为三类:基于Transformer的方法、基于扩散的方法以及基于流匹配的方法。此外,研究者还提出了基于输入优化的方法,通过改进多序列比对、结构模板和输入参数,进一步提升现有预测器的性能。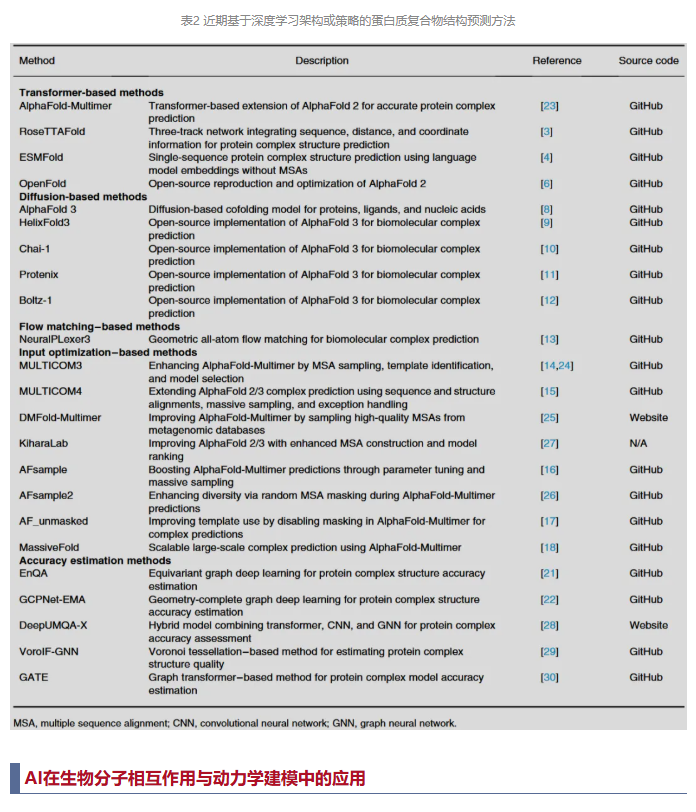
近期CASP16国际竞赛中配体预测类别的一项结论指出,用于蛋白质-配体相互作用预测的新型深度学习共折叠方法(如AlphaFold 3)始终优于排名靠前的基于模板的CASP16预测方法。首次强调在经过盲测评估后,基于人工智能的方法在蛋白质-配体结构预测等方面已超越基于模板的技术。为了更好地理解利用深度学习方法进行生物分子结构、相互作用和动力学建模的最新发展,表3列出了一份精选的方法清单。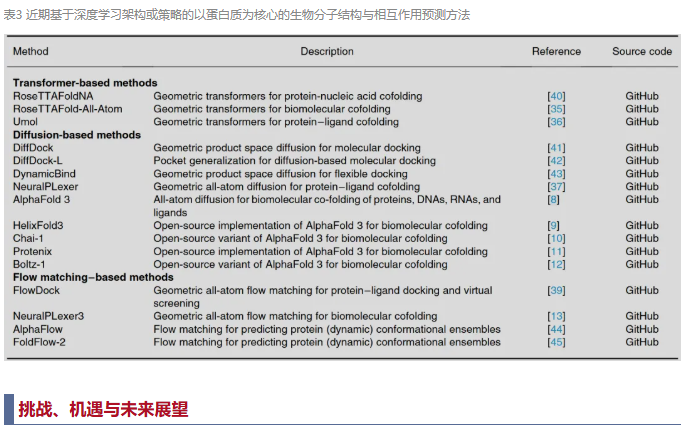
尽管近期已推出多种用于生物分子结构与相互作用预测的深度学习模型,并在方法学上取得了巨大进步,但仍存在若干重大挑战。
首先,现有方法在预测蛋白质-配体相互作用时精度不足,尤其对变构位点预测较差,且模型倾向于记忆模式而非理解底层物理原理。对策是进行算法创新(如神经标度律、精度估计)。
其次,预测具有少量或无同源序列的单体结构仍是难题。解决这一问题需要能利用单序列或有限信息的生成式AI和蛋白质语言模型。
第三,预测由多条链组成的大型蛋白质复合物结构、缺乏链间共进化信息的抗体(纳米抗体)-抗原蛋白质复合物结构,以及非球状蛋白质复合物结构仍然具有挑战性。扩充训练数据集并对现有人工智能方法进行微调可能会进一步改善这一领域。此外,从大量不准确的模型中挑选出准确的结构模型也是一个主要挑战。因此,亟需开发能够可靠且准确评估蛋白质结构模型精度的人工智能方法。
第四,RNA结构预测是AI方法仍落后于传统基于模板或基于分子模拟方法的唯一领域,这也使得预测蛋白质-RNA相互作用充满挑战,主要受限于训练数据不足。除了开发更先进的生成式AI方法外,通过冷冻电镜等实验技术生成更多结构可能有助于缓解这一问题。
最后,蛋白质动力学和替代构象的计算预测仍处于起步阶段。推动该领域发展的一个重要任务是创建大规模、带标注的蛋白质构象集合实验数据集用于训练和测试。
参考链接:
https://doi.org/10.1016/j.sbi.2026.103247
--------- End ---------